比对软件
来啦@-@,今天给各人分享一下几款小编常用的比对软件
Bwa
适用范畴:二代测序数据快速比对到genome上。bwa做为序列比对界的形式软件,短小精悍,适用于多种场所,很有需要搞懂他内部的比对算法,更好也搞懂它是若何实现的。
成立索引:
bwa index in.fasta索引成立好之后会生成五个文件,后缀别离是:
bwt,pac,ann,amb,sa常见比对号令:
BWA-backtrack:illumina reads比对,最长撑持100bp(aln/samse/sampe) bwa aln ref.fa R1.fq > aln1_sa.sai bwa aln ref.fa R2.fq > aln2_sa.sai bwa sampe ref.fa aln_sa1.sai aln_sa2.saiR1.fq R2.fq > aln-pe.sam BWA-SW:long-read比对,长度为70bp-1Mbp;撑持剪切性比对(bwasw) bwa bwasw ref.fa R1.fq R2.fq > aln-pe.sam BWA-MEM:最新,最常用,同SW,但更准更快,与backtrack比拟在70-100bp更具性能优势(mem) bwa mem ref.fa R1.fq R2.fq > aln-pe.samBowtie/Bowtie2
适用范畴:将短序列拼接至模板基因组。
Bowtie在拼接35碱基长度的序列时,能够到达每小时2.5亿次的拼接速度。Bowtie并非一个简单的拼接东西,它差别于Blast等。它合适的工做是将小序列比对至大基因组上去。它最长能读取1024个碱基的片段。换言之,bowtie十分合适下一代测序手艺。
Bowtie2 是将测序reads与长参考序列比对东西。适用于将长度大约为50到100或1000字符的reads与相对较长的基因组(如哺乳动物)停止比对。Bowtie2利用FM索引(基于Burrows-Wheeler Transform 或 BWT)对基因组停止索引,以此来连结其占用较小内存。关于人类基因组来说,内存占用在3.2G摆布。Bowtie2 撑持间隔,部分和双端对齐形式。能够同时利用多个处置器来极大的提拔比对速度。若是目标是与相对较短的参考序列(如细菌基因组)十分灵敏的比对,能够利用Bowtie 2完成,但您可能需要考虑利用NUCmer,BLAT或BLAST等东西。当参考基因组很长时,那些东西可能会十分迟缓,但当参考基因组很短时凡是就足够了。
成立索引:
bowtie2-build genome.fa genome_index bowtie2 -p 10 -x genome_index -1 input_1.fq -2 input_2.fq | samtools sort -O bam -@ 10 -o - > output.bam需要留意的是:genome_index 指的是用于bowtie2的索引文件,而不是参考基因组自己,构建过程参考后文。genome_index 需要指定途径及其共用文件名
RNA-seq比对软件
RNA测序其实不能间接利用DNA测序常用的BWA、Bowtie等比对软件,那是因为实核生物内含子的存在,招致测到的reads其实不与基因组序列完全一致,因而需要利用Tophat/HISAT/STAR等专门为RNA测序设想的软件停止比对。
**01 Tophat2**
能够说是最被公认的RNA测序比对软件(现实上是在DNA比对软件Bowtie的根底上做了一个壳,tophat挪用bowtie,tophat2挪用bowtie2)。
**02 HISAT2**
在Tophat的算法根底了上做了大量的改良,并且克制了Tophat更大的缺点——速度慢(HISAT2既能做RNA-seq也能做DNA-seq)。
①Hisat是一种高效的RNA-seq尝试比对东西
②它利用了基于BWT和Ferragina-manzini (Fm) index 两种算法的索引框架
③利用了两类索引去比对,一类是全基因组范畴的FM索引来锚定每一个比对,另一类是大量的部分索引对那些比对做快速的扩展
成立索引,它的索引就会以genome定名,以*.ht2结尾
hisat2-build -f ref.fasta genome -p 10
而碰到一些大的基因组的时候我们需要用到一个号令参数--large-index,强迫要求产生的索引为‘large’
线程数比对根本的用法:
hisat2 [options]* -x <ht2-idx> {-1 <m1> -2 <m2> | -U <r>} [-S <sam>] hisat2 -x genome -1 ./data/kce/kce_L4_383X83.R2.fastq.gz -2 ./data/kce/kce_L4_383X83.R2.fastq.gz -S test.sam
**03 STAR**
STAR (Spliced Transcripts Alignment to a Reference),用于将测序的 Read 对齐到参考基因组的比对软件,常用于 RNAseq。因其具有较高的准确率,映射速度较其他比对软件高 50 多倍,因而做为 ENCODE 项目标御用 pipeline 东西。它需要占用大量内存,对计算资本有较高的要求。STAR 的默认参数针对哺乳动物基因组停止了优化.
成立索引:
STAR --runMode genomeGenerate \ --runThreadN 50 \ --genomeDir ./hg38_index \ --genomeFastaFiles ./genome.fa \ --sjdbGTFfile ./genes.gtf \ --sjdbOverhang 99参数:
–runMode genomeGenerate:基因组生成形式 –runThreadN:启用线程数 –genomeDir:索引输出途径 –genomeFastaFiles:参考基因组途径 –sjdbGTFfile:参考基因组正文文件 –sjdbOverhang:关于差别长度的读取,抱负值为--sjdbOverhangmax(ReadLength)-1。在大大都情况下,默认值 100 与抱负值类似。STAR 比对:
STAR --outSAMtype BAM SortedByCoordinate \ --runThreadN 20 \ --genomeDir ./hg38_index \ --readFilesIn seq_data_1.fastq seq_data_2.fastq \ --outFileNamePrefix ./seq_data参数:
–runThreadN:启用线程数 –genomeDir:索引途径 –readFilesIn:输入 fastq 的文件途径 –outSAMtype BAM SortedByCoordinate:输出排序的 bam 文件 –outFileNamePrefix:输出文件前缀**04 MapSplice**
TCGA利用的比对软件。
**05 RSEM**
RSEM更像一个软件包而不是一个比对软件,可以供给从比对到计算差别表达的所有步调,因为不需要本身写代码串联差别软件生成的数据格局,因而用起来比力S时S力,值得留意的是,TCGA利用MapSplice比对后再用RSEM计算表达量,并没有间接只用RSEM原拆的Bowtie的比对成果。
①比对软件(RNA-SEQ比对到参考基因组上):bowtie2 tophat hista2 star
②不需要比对(伪比对,基于成立好的参考转录组):sailfish kallsto salmon
③组拆与定量:stringtie cufflink RSEM
性能比力:
①tophat与Hista2算法类似,但是tophat已经不再维护,且比对速度方面,Hista2 显著高于 tophat。STAR与Hista2类似。
②Ballgown在差别阐发方面比cuffdiff更高的特异性及准确性,且时间消耗不到cuffdiff的千分之一
③Bowtie2+eXpress做量量控造优于tophat2+cufflinks和bowtie2+RSEM
长片段比对软件
01 Minimap2
Minimap2是针关于三代测序数据停止比对的东西,minimap2的优势是速度快,并且传闻比对的成果也比力不错;缺点呢,就是消耗内存。根本用法如下:
成立索引:
minimap2 -d co92.min co92.fna比对:
minimap2 -ax map-pb azyz.genome.fasta azyz.flnc.fasta > aln.sam分类:五大类,索引(Indexing),回帖(Mapping),比对(Alignment),输入/输出(Input/Output),预设值(Preset)。
-x :十分中要的一个选项,软件预测的一些值,针对差别的数据选择差别的值 map-pb/map-ont: pb或者ont数据与参考序列比对; ava-pb/ava-ont: 寻找pd数据或者ont数据之间的overlap关系; a**5/a**10/a**20: 拼接成果与参考序列停止比对,合适~0.1/1/5% 序列不合度; splice: 长reads的切割比对 sr: 短reads比对 -d :创建索引文件名 -a :指定输出格局为sa格局,默认为PAF -Q :sam文件中不输出碱基量量 -R :reads Group信息,与bwa比对中的-R一致 -t:线程数,默认为302 ngmlr
NextGenMap-LR(ngmlr)次要用于三代测序的长reads与参考基因组的比对。NGMLR是一款为长reads设想的快速且高精度的停止比对的软件,它是基于NGM开发的,该软件扩展了segmented convex gap-cost scoring model来适应高错误率的长reads比对。
根本用法:
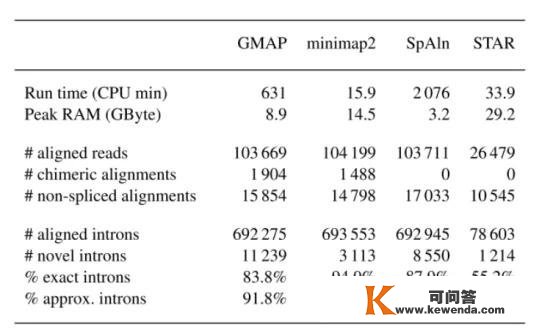
ngmlr -r ucsc.hg19.fasta -q XXX.fastq -o YYY.bam下图是几款软件的比力:
能够看出minimap2表示是非常优良的。