卵白量组学的一个次要目的是供给可以诊断疾病和监测治疗的检测办法。那些检测办法需要对单个卵白量具有灵敏度和特异性,而且在大大都情况下,对统一样品中的一种以上的卵白量停止量化。
目前用于卵白量组学检测的两种次要手艺是基于量谱和亲和分子组合,如抗体。在本文的第一部门,描述了基于那些手艺的最灵敏的现有检测办法,并与ELISA的金尺度停止比力。
阐发灵敏度被定义并与检测限相关,阐发特异性被定义并显示取决于分子校对步调,类似于那些在需要高保实度时应用于生命系统的步调。陈述显示,目前无论是量谱法仍是亲和分子小组都没有供给多重检测所需的灵敏度和特异性的需要组合。
在本文的第二部门,描述了越来越多的利用额外校对步调来连系灵敏度和特异性的检测办法。那些包罗基于临近毗连和慢脱速度润色适体的测定。
最初,本文考虑了在不久的未来可能呈现的改良,并得出结论:进一步开发包罗先辈校对步调的卵白量组学检测办法最有可能供给需要的灵敏度和特异性的组合,而不会产生昂扬的开发成本。
简介卵白量组学是对安康和疾病中卵白量的大规模研究。为了研究的目标,生物体的卵白量凡是被划分为基于差别组织或细胞的卵白量组。然后在尝试中研究那些卵白量组中的卵白量,尝试范畴从旨在识别大量无成见的卵白量组的发现卵白量组学,到旨在量化预选的卵白量组的检测。
从发现卵白量组学到检测,有一个趋向,即从高含量(大量的卵白量被识别)和低灵敏度,到低含量和高灵敏度,本文将讨论那一趋向中的高灵敏度的一端。
血浆卵白量组是本文中描述的卵白量组学检测的次要目的,因为它的可及性,也因为其他卵白量组脱落到血浆中的卵白量被认为能够诊断身体其他部位的安康情况,但那招致的复杂性带来了庞大的挑战。
血浆可能包罗大大都人类卵白量以及来自其他来源的卵白量,如细菌、病毒和实菌[1,2]。那些卵白量的大部门(99%的量量)是以22种高品貌卵白量的形式存在,就血清白卵白而言,其浓度高达10 mM(50 mg/ml),但其他卵白量,包罗一些经常需要测定的卵白量,其浓度要低得多。
最常引用的浓度范畴是从血清白卵白到IL-6(目前诊断检测中含量起码的卵白量之一,在安康人中的浓度约为100 fM)的十个数量级范畴,但许多可能对早期疾病有诊断感化的卵白量可能以更低的浓度存在。
大约22,000个非冗余卵白量(基因产物)的根本数量因为存在许多变体而得到扩展,那些变体可能具有显著的分子类似性,但因为翻译后润色而具有差别的功用。大大都卵白量在生化收集和路子中阐扬感化,凡是是做为多分子复合体的构成部门。
因而,一个卵白量的活性不只取决于其本身的浓度,并且还取决于与之彼此感化或按捺的其他卵白量的浓度。
一个有据可查的例子是细胞因子[3,4]。那个小信使卵白家族参与了多种生化过程,如细胞生长、细胞分化、组织修复和重塑,以及免疫反响的调理。它们经常与其他细胞因子一路阐扬其感化,因而关于单一细胞因子的信息不太可能提醒病人的安康情况。
因而,卵白量组学检测必需可以量化可能以低浓度存在于样品中的多种卵白量,而不遭到可能以更高浓度存在的其他卵白量的干扰。
胜利实现那一目的的检测办法被称为灵敏度和特异性,此中灵敏度是权衡检测办法在检测低浓度目的卵白方面的胜利水平,特异性是权衡它在区分目的卵白和样品中存在的其他卵白方面的胜利水平(那些灵敏度和特异性的定义不该该与诊断灵敏度和诊断特异性相混淆)[5]。
一些卵白量的浓度在**下可能会有多达四个数量级的变革,因而任何检测的另一个重要特征是动态范畴,那是权衡目的卵白量可以被准确量化的浓度范畴。
目前用于检测卵白量组的两种次要手艺是量谱法(MS)和多重免疫检测。本文比力了基于那些手艺的检测办法的长处和缺点,并研究了它们在多大水平上可以供给所需的灵敏度和特异性的组合。
量谱卵白量组学许多版本的MS已经被用于卵白量组学[6-8],但大大都涉及将卵白量消化成肽,然后用HPLC、气相色谱或毛细管电泳停止分馏。
分馏后的肽被送入量谱仪,量谱仪有三个次要部门:电离源、阐发器和检测器。肽被电离并提取到阐发器中,在那里按照它们的量量/电荷(m/z)比率停止别离和检测。随后,通过与肽数据库的比力,对原始卵白量停止离线判定。
最简单的非目的版量谱能够在大约1小时的时间范畴内识别数百种卵白量,但许多卵白量无法检测,因为它们的存在被更丰硕的卵白量所掩盖。因而,量谱的一个次要趋向是开发手艺,通过削减其他卵白量引起的布景信号来进步灵敏度。
一些手艺通过减去样品中的干扰分子来削减布景,同时允许目的卵白或肽流向检测器[9]。减法手艺的例子有HPLC、凝胶电泳和免疫亲和力耗竭。
通过考虑一个详细的例子能够看出减法的效果。
图1 | 消耗丰硕的卵白量对量谱阐发的灵敏度的影响。
(A)全血浆的多重反响监测阐发。
(B)对全血浆停止免疫亲和力耗尽丰硕卵白量的多重反响监测阐发。
(C)无IA消耗的离子电流图,凝胶卵白的峰值用红色暗影暗示。
(D)用IA耗尽20种丰硕的卵白量的离子电流图,凝胶卵白的峰值用蓝色暗影暗示。
缩略语:D,样品的酶解;I,肽的电离;IA,免疫亲和;LC,液相色谱;Q1,阐发仪1;Q2,碰碰池;Q3,阐发仪2。
MRM(多反响监测,也被称为选择性反响监测)版本的MS是在一个串联量谱仪中停止的,它由两个阐发器构成,被一个碰碰池离隔,如图1A所示。来自多达100个消化的目的卵白的特征离子在第一台阐发器中被选择,并在碰碰池中被朋分。
在检测前,那些碎片在第二台阐发器中被别离,成果以总离子电流图的形式呈现,如图1C所示。
图1B显示了MRM的工做流程,此中有一个额外的子牵引步调,即用免疫亲和柱连系血浆中20种丰硕的卵白量;响应的离子流图显示在图1D。比照图1C和D能够看出,因为额外的减法步调削减了布景信号,对应于目的卵白(凝胶卵白)的峰相对更大、更明显。
用免疫亲和力减去丰硕卵白量的MRM的动态范畴约为四个数量级,灵敏度约为10 ng/ml(相当于分子量为50 kDa的卵白量的200 pM),那关于检测许多低品貌的卵白量来说不敷灵敏。
所有的减法手艺城S稀释样品,也可能会非特异性地减去目的分子,因而,应用一种以上的减法手艺其实不必然能进步灵敏度。然而,比来在15 μl血清中检测到了低pmoL浓度的前列腺特异性抗原,其工做流程是先用高pH值的反相HPLC,再用低pH值的反相HPLC和MRM[10]。
那种战略(被称为PRI**)的一个缺点是,即便在低反复程度下,每周也只能检测50个样本。样品产量低是基于MS的卵白量组学检测的一个配合特点;一般来说,在必然时间内能够检测的样品数量跟着灵敏度的进步而削减。
替代减法手艺的一种办法是通过从样品中提取目的分子来削减布景[11]。
提取办法包罗那些提取整类卵白量的办法,如凝固素亲和色谱法,以及基于抗体提取目的卵白量或肽的办法。一般来说,萃取手艺比减法手艺有更高的灵敏度,因为它们浓缩了目的分子而不是稀释了它们,但在最灵敏的时候,它们与基于亲和分子的卵白量组学检测间接合作。
ELISA,卵白量检测的金尺度亲和分子与具有互补的外形和化学性量的目的物量连系。到目前为行,抗体不断是卵白量组学中利用最普遍的亲和分子。抗体连系位点和响应的目的位点之间的平衡能够暗示为:
(1)
此中[B]和[T]是未被占用的抗体连系位点和未被连系的目的位点的摩尔率,[B:T]是被占用的连系位点的摩尔率。公式1中ka和kd是连系息争离的速度常数。连系息争离速度常数的比率被称为抗体的平衡常数或亲和力常数(Ka)。
(2)
高亲和力的抗体要么与目的物量连系得更快(高ka),要么与目的物量连结更长时间的连系(低kd),或者同时具有那两种特征。
基于抗体的卵白量组学检测(免疫检测)能够分为两种形式,取决于成果是通过丈量被占据的仍是未被占据的连系位点来确定[12]。前者被称为试剂溢出型免疫检测,后者被称为试剂限造型免疫检测。
试剂限造型免疫检测一般用于检测那些太小而不克不及包容一个以上的抗体连系位点的物量。免疫合作法检测是试剂限造性免疫检测的例子。
试剂溢出型免疫检测凡是是用两个与统一目的物量上的差别连系位点(表位)连系的抗体来停止的,免疫夹心法是试剂溢出型免疫检测的例子。
那两种形式都能够用一种酶做为标签,但是当人们把ELISA称为卵白量组学检测中灵敏度的金尺度时,他们说的是试剂溢出型的免疫夹心法。
ELISA是指酶联免疫吸附试验,但利用其他标签的免疫夹心法检测,如镧系螯合物[13]、吖啶酯[14]和钌系螯合物[15],也有类似的灵敏度。那些单一目的物量的免疫检测凡是是在96孔或384孔的多孔板中停止的,图2A显示了一个简单的ELISA的次要步调。
图2 | ELISA,卵白量检测的金尺度
(A)酶联免疫吸附夹心法的关键步调(解释见注释)。
(B)酶产品的数量与连系的目的物量的数量成反比,而目的物量的数量又与原始样品中目的物量的数量成反比。
(C)能够按照一系列含有已知浓度的目的物量的校准溶液的反响(可检测的产品量)来构建校准图。红色的高斯曲线是基于零校准液的尺度误差(s)。LOD能够通过在零号校准液的3 × s处程度投影曲到与反响线订交来确定,然后将交点向下垂曲投影到X轴上。在那个例子中,LOD是1.16。
缩略语:E,酶标;LOD,检测限;P,酶催化反响的产品;S,酶底物。
在步调1中,样品与固定在板孔中的捕捉抗体停止孵化。在步调2中,样品被取出,孔被清洗。在步调3中,将带有酶标识表记标帜的检测器抗体参加到孔中,并停止第二次孵化。在步调4中,去除未连系的检测器抗体,并清洗该孔。在步调5中,参加底物并停止第三次孵化,此中酶催化底物转化为可检测的产品,如步调6中所示。
如图2B所示,产品的数量与原始样品中目的物量的数量成反比。凡是情况下,ELISA需要1至2小时才气完成,其动态范畴为三个数量级。
图1B和图2A之间的比力申明了减法和萃取法之间的关键区别:在减法(图1B)中,不是目的物量的物量被从样品中提取出来,而目的物量继续流向检测器,但在萃取法(图2A)中,目的物量被提取出来,样品的其余部门被丢弃了。
抗体微阵列与ELISA的灵敏度多重一词有多种含义,但在本文中,除非另有解释,它是指在不异的未朋分的样品体积中检测一种以上的目的物量。在多重免疫检测的早期,人们遍及认为高重数(高含量)的测定将通过扩大现有的单分子办法来开发。
多重免疫阐发最常见的办法是将差别目的物量的抗体限造在被称为微阵列的微米大小的矩形网格中的已知位置[16-19]。
第一个抗体微阵列与DNA微阵列差不多同时呈现。从那时起,DNA微阵列的开展获得了严重停顿,并获得了相当大的贸易胜利。如今,贸易化的DNA芯片已经能够用于突变阐发、表达阐发和SNP检测,年销售额达数亿美圆。
在开发抗体芯片方面的手艺前进则更为有限。多重免疫夹心微阵列的开展(图3A)被开发统一目的物量上差别表位的配匹敌体的成本所障碍,那种成本跟着测定内容的增加而成倍增加。
图3 | 用于检测卵白量的三种微阵列类型。
(A)已完成的免疫夹心微阵列:工做流程与ELISA类似,但标签必需产生一个不扩散分开其在网格中的位置的信号。
(B)开发抗原捕捉微阵列的次要步调:(1)在平面基量上打印抗体微阵列;(2)标识表记标帜样品中的分子;(3)将标识表记标帜的样品与微阵列孵化;(4)获取完好微阵列的图像。
(C)开发反相微阵列的次要步调:(ⅰ)在平面基量上打印样品;(ⅱ)将特定目的物量的标识表记标帜抗体与微阵列孵化;(ⅲ)获得完好微阵列的图像。
开发单一表位的抗体的成本要低得多,因而大大都的多重免疫微阵列是基于每个目的物量的一个抗体。
那些单抗体免疫微阵列的两个次要形式是抗原捕捉微阵列(图3B),此中样品被标识表记标帜,并用抗体微阵列停止检测;以及反相微阵列(图3C),此中固定肽、抗原、卵白量、细胞或组织样品的微阵列用标识表记标帜的抗体停止检测。在大大都情况下,多重免疫微阵列是在平面基量长进行的,但也能够用编码颗粒的悬浮微阵列停止[20,21]。
一种检测办法的阐发灵敏度是权衡其区分统一物量的差别浓度的才能,但凡是那必然义被更狭义地解释为区分含有和不含有该物量的样品。那种狭义的解释凡是被称为测定的检测限(LOD)。更正式的定义是,LOD是指目的物量不被检测到的概率α(阴性成果)的浓度[22]。
最普遍选择的α值是0.015,它相当于零校准器(已知没有目的物量的样品)尺度误差(s)的三倍。若是α的值为0.015,那么LOD能够通过在Y轴上程度投影零校准器的m + 3 × s(此中m是均匀反响)的点,曲到它与最合适的剂量-反响线订交,然后将交点向下投影到X轴,如图2C所示。
LOD的可靠性取决于它所基于的丈量数量,因而用于确定LOD的反复数(n)也应该被陈述;关于普遍利用的95%的置信区间来说,至少n = 6是能够承受的。在严酷开发的免疫夹心微阵列中,s的大小次要取决于非特异性连系的数量和跟着测定的停止而积累的误差。
若是假设标签具有无限的特异性活性,并对非特异性连系量和误差停止假设,就能够用公式3来估量试剂残留免疫阐发的LOD:
(3)
此中非特异性布景信号(N**)长短特异性连系量占标识表记标帜的检测器抗体总浓度的一部门,CV是与零校准物相关的相对误差(s/m),Ka2是检测器抗体的平衡常数或其等价物[23]。留意N**在那里是决定灵敏度的关键因素,就像在MS中一样。
图4显示了若是假定N**和CV的值为0.001和0.01,免疫夹心微阵列的灵敏度若何取决于Ka2。0.001的非特异性连系和0.01的相对误差是在理论中能够实现的低端,而优良的阐发级卵白量单克隆抗体的Ka值在1×10^10摆布。
图4 | 免疫夹心法检测的检测限取决于检测器抗体对连系目的物量的亲和力常数。
缩略语:LOD,检测限。
因而,图4预测免疫夹心微阵列的灵敏度将集中在1×10^-15 mol/L的浓度上。因为TSH在临床上的重要性,良多人都努力于开发TSH的免疫夹心微阵列。对贸易来源的TSH免疫微阵列的审查发现,其灵敏度在0.005 ~ 0.1 mIU/L之间[24]。
若是假设人类TSH的活性为5 IU/mg,MW为28 kDa,那相当于灵敏度在35 ~ 714 × 10^-15 mol/L之间。如图5所示,对免疫夹心微阵列的陈述LODs的普遍查询拜访表白,它们集中在50 × 10^-15 mol/L的尺度浓度上。
图5 | 免疫夹心法(ELISA和时间分辩荧光)的检测限陈述。
缩略语:LOD,检测限。
那些对灵敏度的估量适用于试剂溢出型检测,在那种检测中丈量被占领的连系点,如免疫夹心法检测。丈量未被占据的连系位点的亲和力办法(如免疫合作法检测)的灵敏度较低,因为它们涉及到在大布景下检测一个小信号。
理论模子表白,那品种型的检测办法的灵敏度能够通过亲和力常数(Kd)的倒数乘以相对误差(即CV × Kd)来估量[25]。因而,利用Kd = 1 × 10^-10和CV = 0.01抗体的免疫合作法检测LOD将是1×10^-12 mol/L。
在理论中,免疫合作法检测次要用于检测低分子量的分子,关于那些分子,抗体的Ka值凡是低于1 × 10^10,因而灵敏度也响应较低。
经常有人声称,抗体捕捉微阵列免疫检测的LODs在pM-fM范畴内具有灵敏度(可与ELISA相媲美)[26-30],但是没有统计学上的可靠成果来撑持那一说法。
在此中一项较好的研究中,抗原捕捉微阵列是通过将8种补体卵白的单链抗体连系位点装点在黑色聚合物微阵列玻片上而造备的[31]。微阵列与用生物素标识表记标帜的血清和血浆样品停止孵化,然后用荧光链霉菌素标识表记标帜。
通过减去每个点的部分荧光来确定反响,以抵偿布景信号的部分变革,并利用一个内部对照来使差别微阵列的反响一般化。每个卵白量的两个更高和更低的反响被排除,LODs是基于其余四个反复(n = 4)。
八种补体卵白的LODs(基于2 × s的零校准物)在0.35 ~ 530 pM范畴内(八种目的细胞因子的均匀LOD=10^7 pM),那比ELISA的灵敏度低三个数量级,但比MRM版本的MS更灵敏。
公式3显示,决定灵敏度的一个关键因素是布景信号的大小。在免疫夹心微阵列中,布景信号次要是因为非特异性连系的标签形成的,那取决于所用的标识表记标帜抗体的浓度(图2A的步调3)。在抗原捕捉微阵列中,样品中的所有分子都被标识表记标帜,因而它们都有可能招致非特异性连系。
因而,公式3中的N**关于抗原捕捉微阵列来说不成制止地要高得多,因而它们的灵敏度低于ELISA和其他设想优良的免疫夹心微阵列也就层见迭出。反相微阵列可能比抗原捕捉微阵列更灵敏,因为N**取决于标识表记标帜的抗体的浓度,而不是样品中所有标识表记标帜的分子。
特异性的来源,分子校对步调一个抗体的阐发特异性是权衡它在指定的目的物量和样品中存在的其他物量之间区分的有效水平。与抗体连系的其他物量被称为穿插反响物。特异性取决于穿插反响物的相对浓度和亲和力。
高浓度的穿插反响物能够扭曲用抗体检测低品貌目的物量的成果,即便目的物量的Ka比穿插反响物的亲和力常数(Kac)高良多。缺乏特异性仍然是障碍开展多重免疫微阵列的次要障碍之一。
免疫检测中的各个步调能够被看做是一系列的分子校对步调,招致误差分数(穿插反响物与目的物量的比率)的逐渐削减。
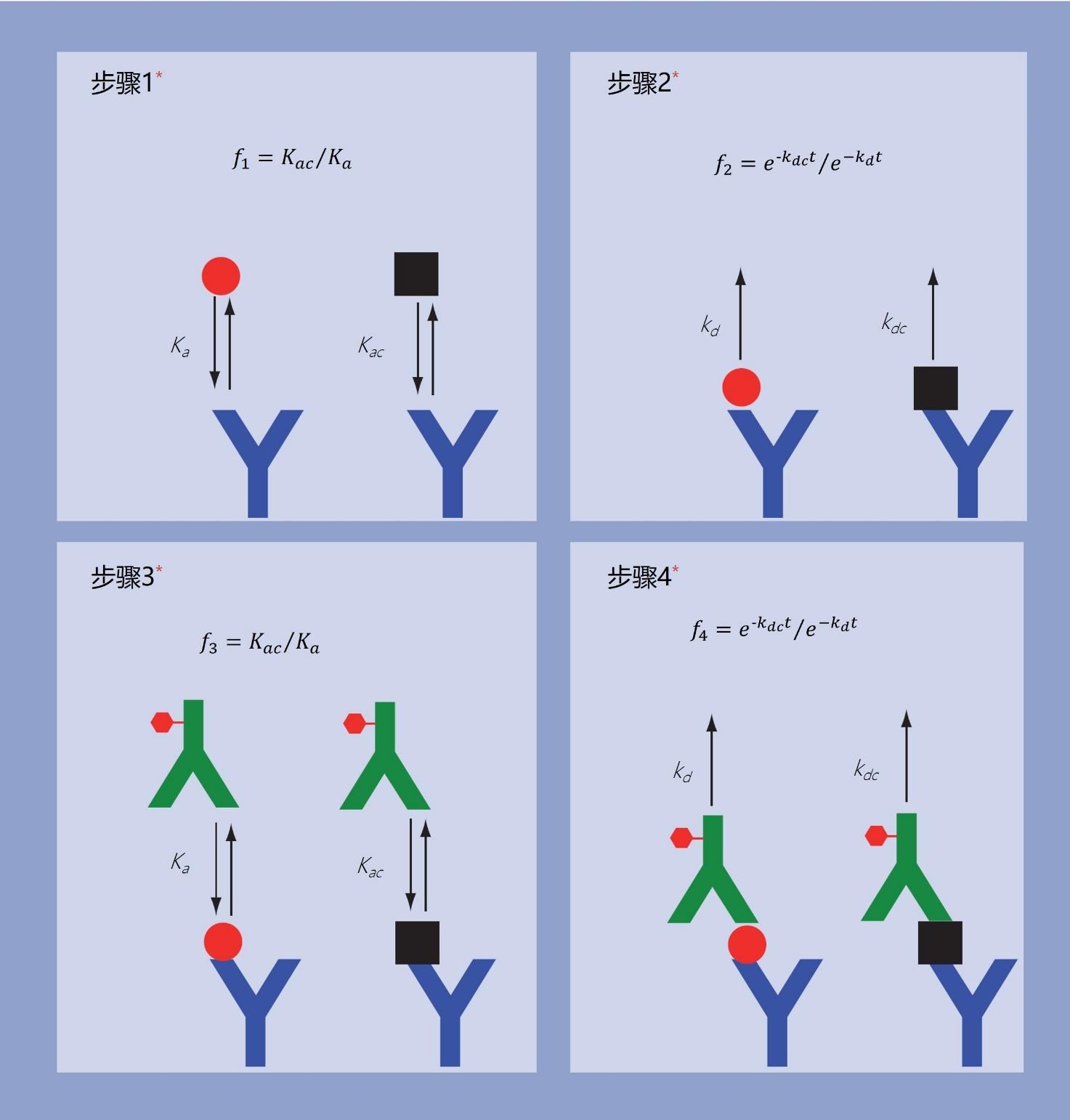
图6 | 免疫夹心法的四个阶段被视为校对步调,此中错误分数(预定的目的分子[红色圆圈]与穿插反响物量[黑色方块]的比率)被持续削弱。插图中的公式显示了若是只要一种穿插反响物量,每个步调的误差分数将若何计算。
图示:每个红色星号对应于一个校对步调。
图6显示了双位点免疫检测中每个步调的误差分数(f)。图6中显示的f的插入公式是基于以下假设:步调1和3到达平衡;步调2和4的持续时间为t秒;步调1中抗体的浓度小于(0.05/Ka);步调1中目的物量和穿插反响物量的浓度不异,且小于(0.001/Ka)。免疫检测的总体误差分数(ft)为:
表1显示了若是Ka = 10^10,Kac = 10^8,kd = 10^-5/s,kdc = 10^-3/s和t = 300 s,免疫检测中每一步的f和f值。按照那些值,四步后的累积误差分数ft比两步后的ft小两个数量级。那品种型的计算表白为什么两步抗原捕捉微阵列的特异性低于四步免疫夹心法检测。
表1 | 基于Ka 10^10,Kac 10^8,kd 10^-5/s,kdc 10^-3/s和t 300 s的简单免疫夹心法检测中每个步调的误差分数(f)和累积误差分数(ft)
步调fft步调11.0 × 10^-21.0 × 10^-2步调27.4 × 10^-17.4 × 10^-3步调31.0 × 10^-27.4 × 10^-5步调47.4 × 10^-15.5 × 10^-5虽然灵敏度和特异性较低,但由数百种抗体构成的抗原捕捉微阵列在研究中被普遍利用。一个常见的应用是生物标记物的发现,那涉及到用抗体微阵列比力安康(对照)和疾病(病例)样本的成果[32-36]。
凡是情况下,那些微阵列是用贸易来源的抗体系体例备的,用于定名目的物量。Loch等人利用由320个整体单克隆和多克隆抗体构成的抗原捕捉微阵列来比力卵巢癌病例和对照组的血清样品[36]。
一些目的物量用一种以上的抗体停止了检测。例如,卵白量CA125(一种已知的卵巢癌生物标记物)被用八种差别的抗体检测,那些抗体给出的不克不及区分癌症患者和对照组的概率分数(不异队列大小的P值)从0.003到0.171不等,错误发现率(FDRs)从0.214到0.433(即每100次检测,错误阳性的数量在21到43之间,取决于抗体的选择)。
那些庞大的变革表白,用抗原捕捉微阵列获得的成果可能在很大水平上取决于抗体的选择,而不是病例和对照之间的任何现实差别。
抗原捕捉微阵列的利用是基于抗体对单一目的物量具有特异性的假设,但是十多年前,人们发现115个微阵列的抗体中只要20%对其目的物量产生了定量的准确成果[37]。
比来,当用酵母卵白的反相微阵列研究11种贸易来源的抗体的特异性时,发现有5种抗体对其目的物具有特异性,5种抗体与多种抗原发作穿插反响,1种抗体与1000多种差别的抗原连系[38]。
那些成果表白,许多单个抗体的特异性不敷以识别其指定的目的物量,并有助于解释为什么很少有生物标记物研究被转化为临床试验。
MS与亲和力分子的连系,灵敏度/特异性问题的处理计划进步卵白量组学检测的特异性的一种办法是将抗原捕捉与量谱连系起来。那种组合被称为不变同位素尺度与抗肽抗体捕捉(SISCAPA),被Whiteaker等人用来检测血浆中的9种卵白量,如图7所示[40]。
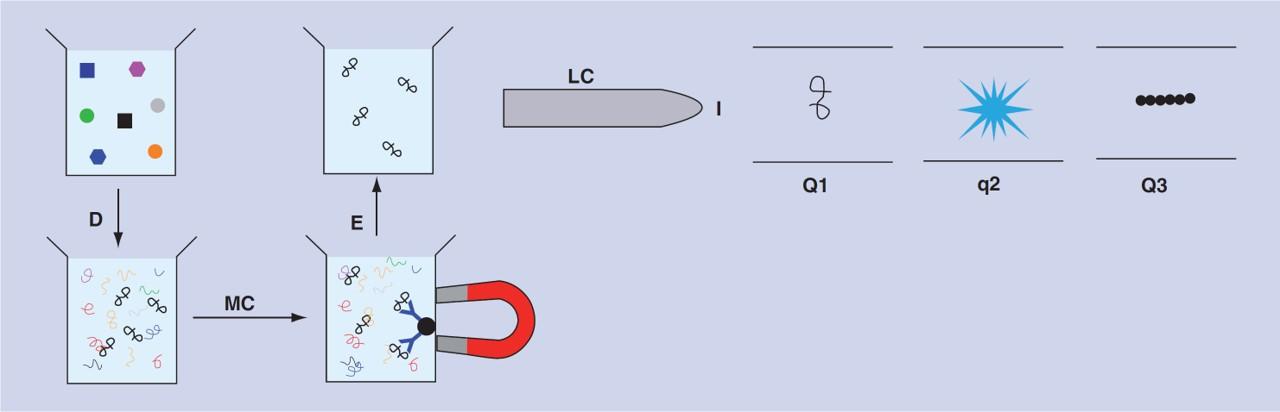
图7 | 用抗肽抗体捕捉的不变同位素尺度,抗原捕捉和量谱阐发相连系。
缩略语:D,样品的酶解;E,肽与抗体的解离;I,肽的电离;LC,液相色谱;MC,与抗体连系的肽的磁捕捉;Q1,阐发仪1;Q2,碰碰池;Q3,阐发仪2。
用胰卵白酶消化样品(每种卵白量检测10 μL血浆),然后用抗体和磁珠捕捉每种目的卵白量的一个肽。选择肽是因为它们是目的卵白所特有的,而且在MS中表示优良,但不是因为它们是抗体的好目的。
从抗体中洗脱出来的多肽用液相色谱法停止分馏,并利用MRM办法停止MS检测。一起头有15种卵白量被做为目的,但后来有6种因为在抗体捕捉阶段收受接管率低(<7%)而被放弃。其余检测的特异性很高,因为MS被用做高保实校对步调以及检测。
检测的动态范畴为三到四个数量级,均匀LOD(m + 3 × s)为88 pM,那与抗原捕捉微阵列类似,但比ELISA低两到三个数量级。通过从更大致积(1 mL)的样品中提取多肽,LODs被降低到3 pM,但在许多情况下不会有那么大致积的样品。
虽然LODs与抗原捕捉微阵列相当,但后者的多重版本具有不那么复杂的工做流程和更快的吞吐量,而且需要更小的样品量。
SISCAPA与ELISA的比力表白,因为在抗原捕捉阶段多肽的不成反复的收受接管而招致的禁绝确,但那可能会被一个不那么复杂的工做流程所改善。
将抗体捕捉的灵敏度和量谱的特异性连系起来,听起来像是对灵敏度/特异性问题的处理计划,但一个检测办法只要在其最单薄的环节才是好的,而SISCAPA的单薄环节是量谱的灵敏度。
因而,那一范畴的停顿将取决于量谱灵敏度的进步,但要与下面描述的基于亲和分子的多重检测合作,必需填补三到四个数量级的差距,那在不久的未来是不成能被逾越的。
系统产生更多亲和力分子利用抗原捕捉微阵列而不是免疫夹心微阵列的一个常见原因是没有针对统一目的物量的差别表位的婚配抗体对。对更普遍的廉价亲和试剂的需求是几个大型开发项目标主题。
人类卵白量图谱项目已经开发了一个以基因为中心的管道,用于系统生成和验证亲和纯化的多克隆抗体[41,101]。那条流水线的起点是在计算机中识别与其他卵白量同源性低的80 ~ 100个氨基酸表位标签。那些标签被表达为重组卵白片段并用于在兔子体内培育抗体。
用不异的表位标签做为配体,通过亲和层析法纯化抗体,并用肽的反相微阵列停止验证。
若是一个抗体的大部门连系是针对准确目的的抗原,那么它就被认为是有效的。我们的目的是针对统一卵白量上不堆叠的表位产生至少两个验证的抗体。
当下,已经有超越34000个抗体被验证,而且新的抗体正在以每月492个的速度增加,但是那些数字是基于一个十分弱的验证形式。在更严酷的Western Blot验证中,22,000个测试的抗体中只要531个抗体与预期的MW的单一条带连系[42]。
一些额外的条带可能是因为统一卵白量的变体或复合物形成的,但是那一点还没有被证明。人类卵白量图谱中颁发的所有抗体都能够在S场上买到,但是到目前为行,还没有任何抗体被转化为有效的诊断测试。
亲和卵白量组专注于利用高通量体外选择办法做为增加可用亲和试剂范畴的手段[43,102]。全长抗体中只要一小部门参与了与目的分子的连系。那一部门能够通过噬菌体和核糖体展现等办法,在体外表达为与编码它的DNA相关的肽。
然后,通过对多肽组合库停止频频的亲和力捕捉和编码DNA的扩增轮回,就能够判定重组抗体。那个过程比在动物身上消费抗体的办法要快(几周而不是几个月),并且成本较低,因为它能够在小体积的溶液中停止,需要的根底设备较少。
也有可能为那些对动物宿主有毒或因与动物本身抗原类似而无法产生反响的目的消费重组抗体。体外办法还能够包罗一些前提,以进步对目的分子的细小改动(如翻译后润色)的识别才能,或削减对可能的合作敌手的穿插反响[44,45]。
在某些情况下,体外选择被用来进步重组抗体的Ka值,使其超越体内施加的大约10^10的上限[46,47],但进步亲和力其实不必然能进步特异性。大大都重组抗体的亲和力可与动物体内产生的抗体相媲美。
包罗更多校对步调的特异性基因分型检测法缺乏特异性其实不局限于基于抗体的检测。它对核酸检测的影响在单核苷酸多态性(SNP)的检测中最为明显,在那种检测中,有需要对仅有一个碱基差别的序列停止区分[48]。
明白的核酸化学允许在严酷的前提下通过将核酸序列与固定的寡核苷酸探针微阵列停止杂交来克制那个问题,但每个SNP必需用多个探针停止检测,以区分完全婚配和不婚配的杂交。
例如,由Affymetrix基因分型微阵列判定的每个SNP要用40个差别的探针停止检测[49]。一个由四个探针构成的区块包罗与SNP完全婚配的探针和三个与完全婚配的探针,除了在SNP的基因座上有一个差别的碱基;那些碱基位于探针序列的中间位置。
别的两个由8个探针构成的区块,所有那四个碱基都从序列的中间移到±1和±4个碱基。第四块20个探针由其他20个探针的反义版本构成。通过从完美婚配产生的更强烈的信号中减去穿插反响探针的反响来判定SNP。
卵白量的化学成分比核酸更多样化,因而不克不及用抗体产生相当于基因分型的微阵列,但也能够通过施行一系列校对步调而不是用多个探针检测来检测SNPs。在golden gate检测法[50]中,基因组DNA被生物素激活并附着在链霉菌素磁珠上。
在检测的步调1,磁珠与等位基因特异性探针(ASP)和基因组特异性探针(LSP)一路孵化,前者与基因组目的序列杂交,其3′端与SNP的位置相婚配,后者与ASP的上游杂交,如图8所示。
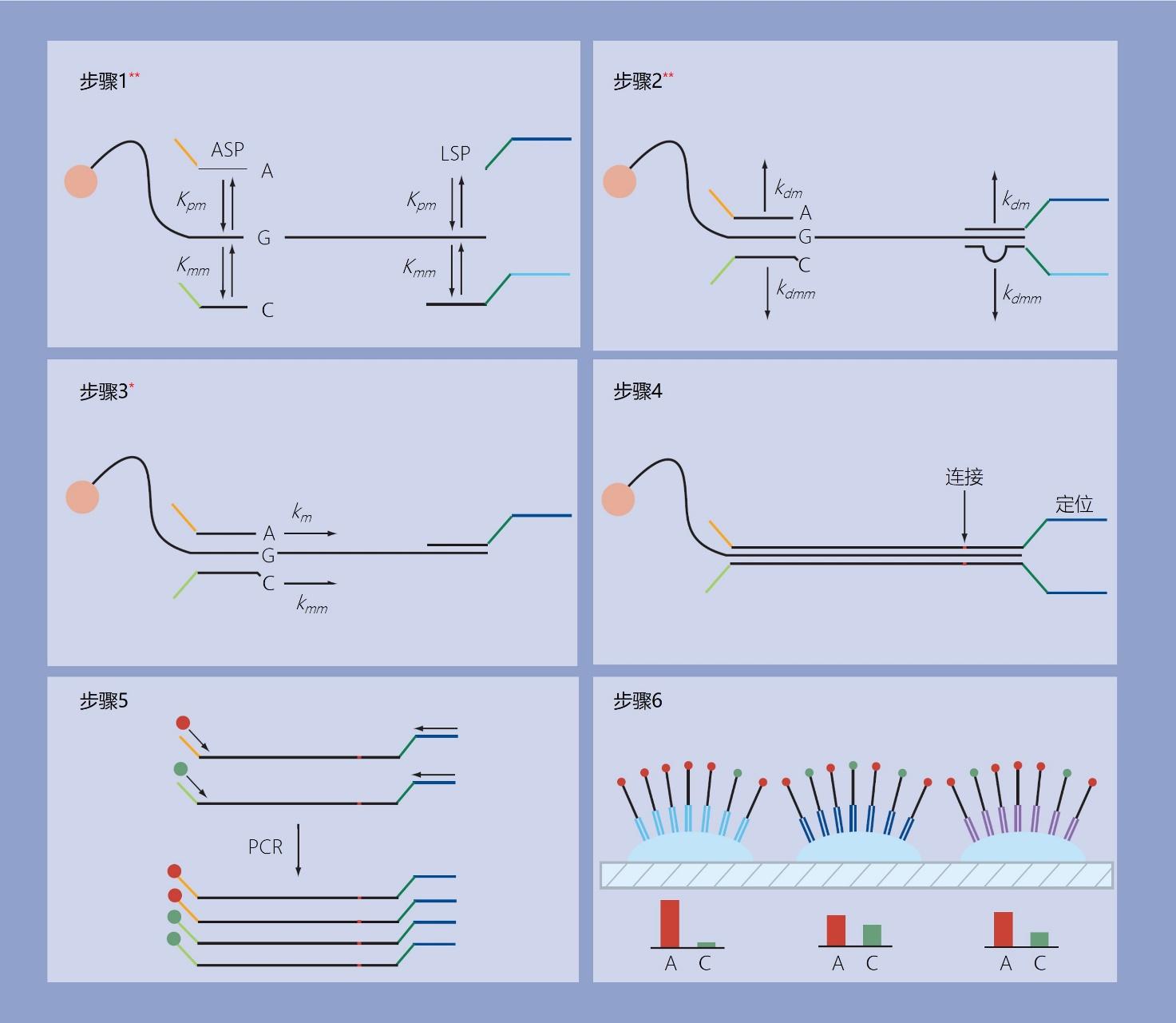
图8 | golden gate基因分型检测,次要校对步调的位置和数量由插入的红色星号暗示。
步调1:ASP和LSP与附着在磁珠上的基因组目的序列停止合作性杂交,那是两个校对步调。
步调2:严酷清洗,优先别离不准确的杂交探针,那是两个校对步调。
步调3:用婚配的3′端碱基耽误ASP,那是一个校对步调。
步调4:将耽误的ASP与LSP毗连。
步调5:用荧光引物对毗连的探针停止PCR。
步调6:将PCR产品与光纤板中的探针微阵列停止杂交。
缩略语:ASP,等位基因特异性探针;LSP,基因座特异性探针。
在步调2中,对珠子停止严酷的洗涤,使不准确的杂交探针别离,在步调3中,用一种对SNP基因座上的错配高度灵敏的聚合酶扩展ASPs。在步调4中,用DNA毗连酶将扩展的ASPs与LSPs毗连起来,在步调5中,利用ASPs和LSPs中的通用PCR位点扩增毗连的探针。
在步调6中,PCR产品与LSP中地址序列互补的寡核苷酸微阵列停止杂交。该检测的前三个步调包罗五个校对步调。那些校对步调中错误分数的累积衰减使得SNPs能够在不利用大量探针的情况下被判定。
如图6所示,前四个校对步调与免疫夹心法检测中的校对步调相当,但没有相当于第五个校对引物延伸步调。那些校对步调类似于付与许多生物高特异性反响动力XX对计划中的步调[51]。
在那些计划中,通过扩大所需成果的自在能变革和非特异性副反响的自在能变革之间的差距,总体误差部门被逐步削弱。只要有高特异性的要求,生物系统就会接纳那种计划,那一察看表白,基于亲和分子的卵白量组学检测的特异性也能够通过增加校对步调的数量和/或严酷水平来进步。
通过增加校对步调进步亲和卵白量组学的特异性在免疫检测中增加校对步调的一种办法是将其与MS利用的一种分馏手艺相连系[52]。Wu等人利用尺寸排除色谱法将生物素标识表记标帜的细胞裂解液合成为10到670 kDa的20个馏分。每个馏分都与300个毗连到编码微珠的抗体停止孵化,然后用荧光链霉菌素标识表记标帜与洗净的微珠连系的抗原,并通过流式细胞仪检测。
做者暗示,一些卵白量在只包罗几千个细胞的样品中能够检测到,但因为没有LOD,所以不成能与其他办法比力灵敏度。只要穿插反响物量不与目的物量在统一馏分中洗脱,特异性必定会进步,但灵敏度可能会跟着样品稀释招致的馏分数量增加而降低。
免疫检测中最出名的附加校对步调的例子是接近毗连[53,54]。在接近毗连免疫检测(PLA)中,抗体被毗连到一个寡核苷酸接近探针上,该探针由一个靠近抗体的奇特识别序列和一个通俗毗连序列构成。
当一个分子被夹在两个抗体之间时,探针被带到接近的处所,在那里与一个毗连的寡核苷酸杂交在热力学上是有利的。
在一个多重检测中,抗体连系反响是在溶液中停止的,但在低温下留宿孵化是为了促进热力学上最有利的杂交。然后探针通过酶连接构成陈述序列,该序列被扩增并通过qPCR检测。
那允许对24种卵白量停止灵敏的多重免疫夹心法检测,而不需要停止大量的抗体选择和优化[55]。
那种无别离版本的PLA的次要缺点是接近探针的浓度必需低,以制止非特异性毗连,因而检测的动态范畴很短[56]。那一限造能够通过利用别离和洗涤步调来克制,如图9所示,如许能够停止额外的校对步调[57]。
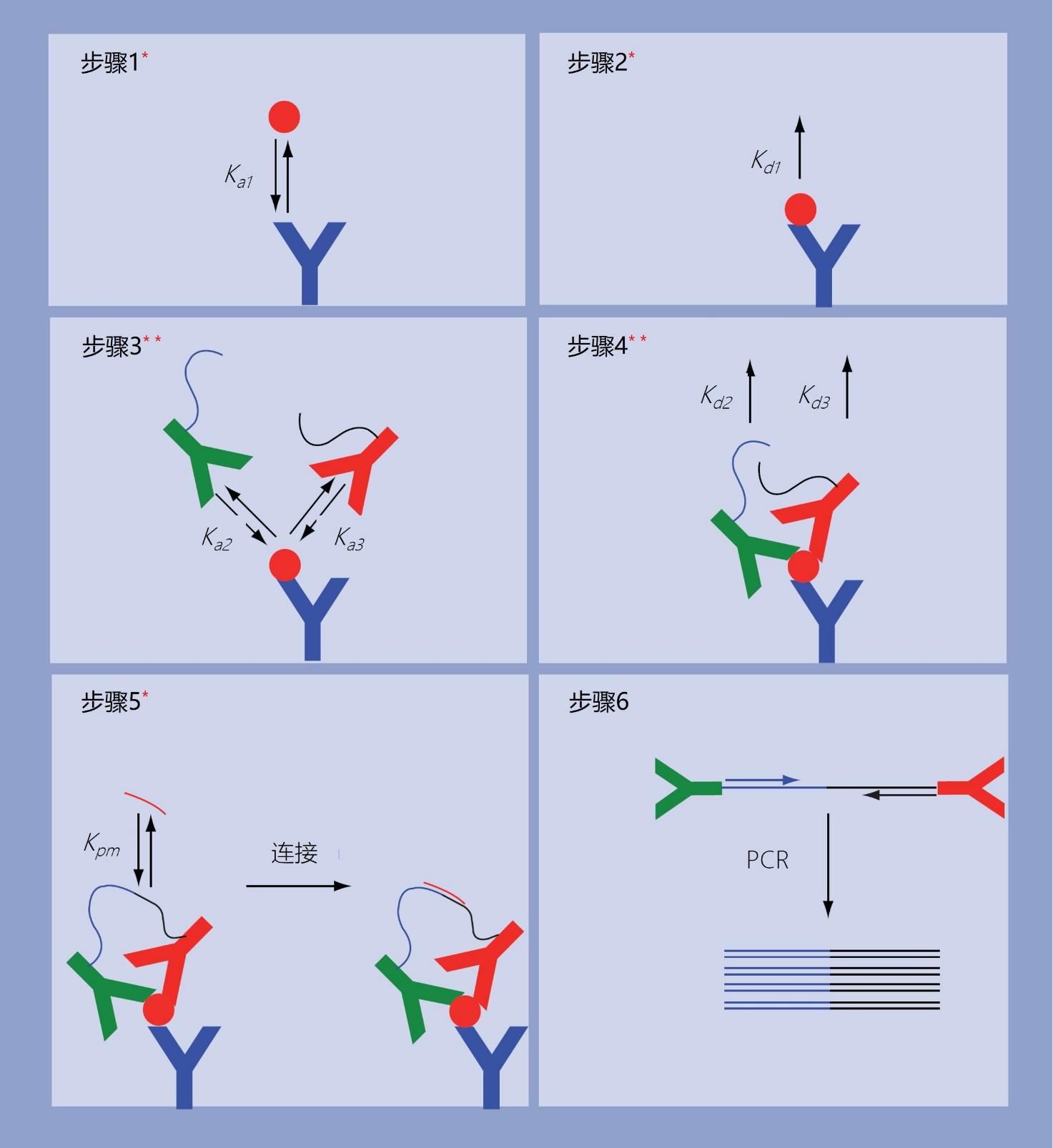
图9 | 在[57]中描述的接近毗连免疫检测,次要校对步调的位置和数量由插入的红色星号暗示。
步调1-4与免疫夹心法检测类似,只是通过利用两个检测器抗体来增加校对步调的数量。检测器抗体用寡核苷酸标识表记标帜,只要在步调5中,若是它们的位置很近,才会毗连在一路。在步调6中,通过实时PCR检测已经毗连在一路的寡核苷酸。
那个版本的PLA的均匀LOD(m + 2 × s)为10 fM,与ELISA的更佳形态相当,而动态范畴为5到6个数量级,效果更好。在图9所示的例子中,通过利用两个检测器抗体,特异性得到进一步加强。还有一种原位版的PLA,用于细胞和组织中的单个卵白量和卵白量-卵白量复合物,毗连后构成一个圆形模板,通过部分滚动圈扩增检测[58]。
第一个关于PLA的陈述部门是基于两个与凝血酶差别表位连系的DNA适配体[59]。适配体是单链核酸,具有与抗体相当的识别特征,因为它们能够折叠成复杂的三维外形,与普遍的目的实体特异性连系[60,61]。
它们是通过挑选10^12-10^16个寡核苷酸的组合库来获得与所选目的分子连系的图案。挑选过程被称为指数富集配体的系统进化(SELEX),包罗体外选择和PCR扩增的频频轮回。
在选择步调中,单链寡核苷酸合作目的物量上的表位,而在扩增步调中,寡核苷酸池被富集告终合的序列。然后,富集的池子被送入下一个选择和扩增周期。凡是情况下,要停止5到20个轮回,然后通过克隆和测序判定剩余池中的适配体。
虽然有良多长处,但适配体还没有被普遍用于卵白量组学检测,因为一般来说它们的亲和力比抗体低(因而灵敏度也低),如图10所示。
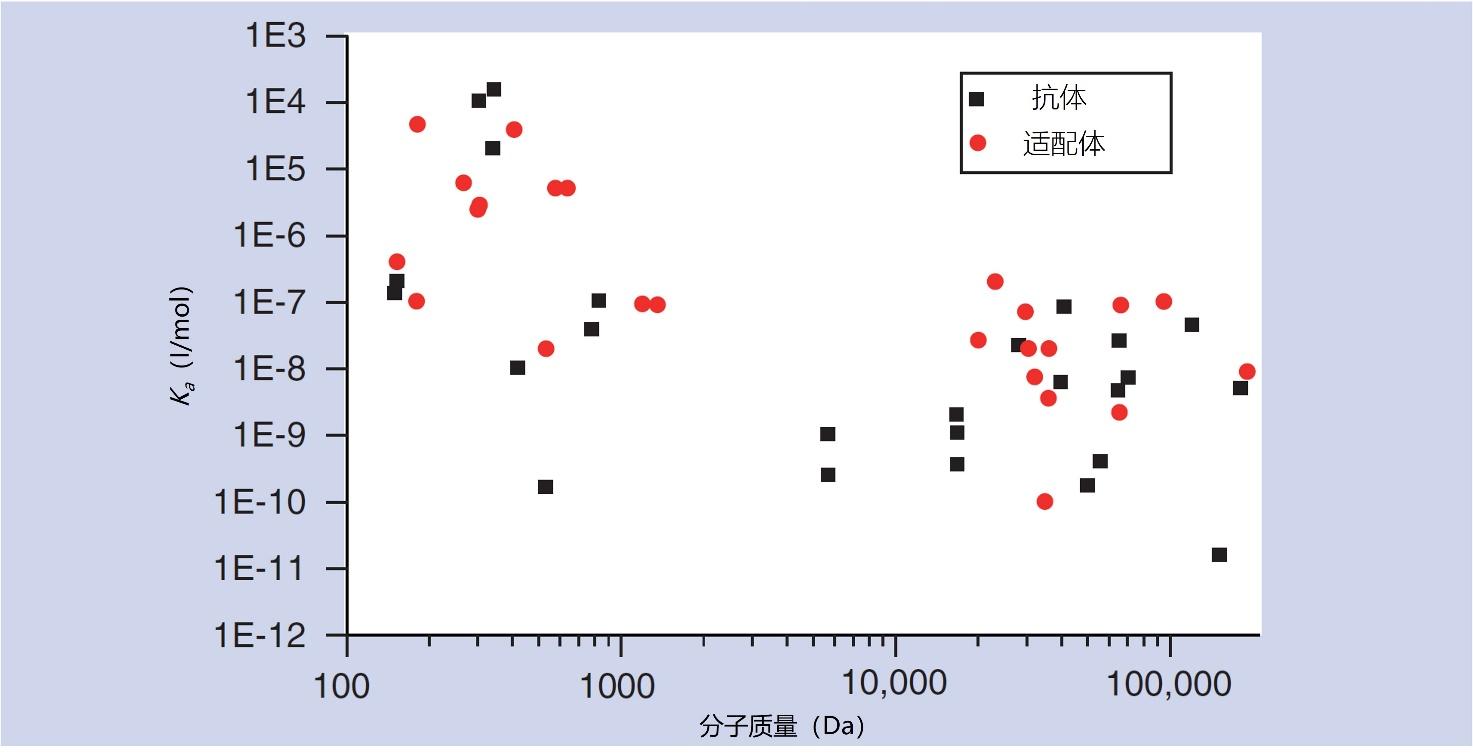
图10 | 抗体和DNA诱导体亲和力的比力,显示了它们若何取决于目的物量的分子量。当目的物量的分子量较低时,DNA适配体的亲和力与抗体相当,但一般来说,对高分子量物量的亲和力不如抗体。
另一个问题是,关于大大都卵白量来说,所有已经确定的合剂都与不异的表位连系,因而无法获得夹心法检测中可能的额外校对步调[62]。像图6所示的校对模子的一个长处是,它们提醒了哪些步调最合适改良。
表1中的模仿值表白,解离步调(步调2和4)比连系步调(步调1和4)在削减错误分数方面的效果要差。比来,Gold等人引入了一类新的合剂,称为慢速别离率润色的合剂(SOMAmers),那些合剂因其与目的分子的迟缓解离率(低kd值)而被选中[63,64]。
那使得亲和力测定中的洗涤步调能够在更严酷的前提下停止,从而招致非特异性连系的诱导剂的优先解离。那就填补了第二个合拍体所供给的额外校对步调。基于SOMAmers的多重检测,其均匀LOD为300 fM(m + 3.2 × s),动态范畴为7个数量级,已被用于识别各类医疗前提的生物标记物,包罗肺癌和慢性肾病[63,65]。
卵白量组学检测的现状和标的目的正如本文题目所示,需要连系灵敏度和特异性的卵白量组学检测办法。基于量谱和抗原捕捉(SISCAPA)的检测办法具有高度的特异性,但它们的灵敏度不敷以检测大大都低品貌的卵白量。
免疫夹心法检测的灵敏度足以检测许多如许的卵白量,但因为识别足够特异的婚配抗体对的成本很高,所以多重测定仅限于高价值的目的物量。
处理灵敏度/特异性问题的一个法子是进步SISCAPA的灵敏度,但灵敏度的差距很大,即便它能够被填补,也很难看到基于MS的检测若何可以在资本丰硕的尝试室以外的处所摆设。
跟着卵白量组学检测的目标从发现转向安康和保健,对能够在资本有限的处所摆设的廉价卵白量组学检测的需求将增加[66]。灵敏度/特异性问题的另一个处理计划是找到进步基于亲和力的多重检测的特异性的办法,而不产生昂扬的开发成本。
基于抗体的多重检测能够摆设在资本有限的处所[67,68],但关于大大都目的物量来说,使其阐扬感化所需的抗体是不成用的。一些项目正在扩大可用的亲和力试剂的范畴,但在开发多重免疫检测时产生的大部门成原来自于选择没有穿插反响的抗体,而不是抗体自己的产生。
因而,只要开发出成本较低的将亲和力试剂纳入多重检测的办法,扩大亲和力试剂的范畴才气招致卵白量组学的停顿。PLA是那个标的目的上的一个重要停顿,因为它降低了将抗体纳入多重检测的成本,但它需要婚配的一对以至三匹敌体[57]。
PLA最后是用诱导剂[59]演示的,但因为到目前为行只发现了一对颠末验证的婚配的诱导剂(用于凝血酶),所以排除了利用那些亲和试剂的进一步工做[62]。那是不幸的,因为PLA与诱导体的组合比抗体更容易实现,而且具有更高的校对潜力。
DNA适配体属于一组分子,是抗体的廉价替代品。那些替代品包罗通过噬菌体和核糖体展现手艺别离出来的多肽适配体[69],但DNA适配体做为最简单和最经济的消费体例脱颖而出。
Gold等人胜利地开发了上述高重数的多重检测办法,原因之一是他们的SOMAmers具有类似氨基酸的功用团[63,70]。那种化学反响是由Eaton等人开发的[71],并构成了一种办法的一部门,那种办**朝着与酶促扩增相适应的类似肽的混合分子的标的目的开展[72]。
为什么只发现了一对婚配的适配体,一个建议是,与基于氨基酸的抗体连系位点比拟,PCR可扩增的核苷酸的低化学多样性凡是排除了婚配对[71,73]。引入更多的与PCR兼容的核苷酸化学成分隔辟了新的设想空间,应该更容易识别除凝血酶以外的卵白量的婚配对合剂。
那将与基于分子生物学东西的高级校对步调的多重卵白量检测相兼容[74]。基于肽类核酸诱导体和更多校对步调的多重卵白量组学将与目前的共识有很大的差别,但希望通过强调现有卵白量组学检测中存在的问题,本文将为那一标的目的的进一步开展供给动力。
将来瞻望对灵敏和特异的卵白量组学检测的需求是MS或传统的抗体微阵列所不克不及满足的。量谱不敷灵敏,传统的抗体微阵列要么不敷特异,要么开倡议来不划算。
卵白量组学检测的抱负灵敏度和基于MS的检测的现实灵敏度之间的差距十分大,在将来5年内不成能被填补,但基于亲和分子的检测的特异性能够通过更好的校对来进步,而不会产生昂扬的开发成本。
有两项开展出格显示了改良校对的优势:带有近似毗连的抗体和SOMAmers。每种办法都有其特殊的优势。在将来五年内,估计连系那些优势的卵白量组学检测办法将起头呈现。
概念总结卵白量组学检测必需具有足够的灵敏度和特异性,以便在其他卵白量存在的情况下对多种卵白量停止量化,那些卵白量可能以更高的浓度存在。基于量谱的卵白量组学检测不敷灵敏。基于抗体捕捉的卵白量组学检测既没有足够的特异性也没有足够的灵敏度。多重免疫夹心法检测的开发成本对大大都应用来说太高。连系亲和分子和量谱的卵白量组学检测不敷灵敏。生物和基因组学检测中高特异性的来源是分子校对。基于亲和分子的卵白量组学检测,包罗额外的校对步调,具有灵敏度和特异性的更佳组合,以及将来开展的更大潜力。概念总结卵白量组学检测必需具有足够的灵敏度和特异性,以便在其他卵白量存在的情况下对多种卵白量停止量化,那些卵白量可能以更高的浓度存在。基于量谱的卵白量组学检测不敷灵敏。基于抗体捕捉的卵白量组学检测既没有足够的特异性也没有足够的灵敏度。多重免疫夹心法检测的开发成本对大大都应用来说太高。连系亲和分子和量谱的卵白量组学检测不敷灵敏。生物和基因组学检测中高特异性的来源是分子校对。基于亲和分子的卵白量组学检测,包罗额外的校对步调,具有灵敏度和特异性的更佳组合,以及将来开展的更大潜力。概念总结卵白量组学检测必需具有足够的灵敏度和特异性,以便在其他卵白量存在的情况下对多种卵白量停止量化,那些卵白量可能以更高的浓度存在。基于量谱的卵白量组学检测不敷灵敏。基于抗体捕捉的卵白量组学检测既没有足够的特异性也没有足够的灵敏度。多重免疫夹心法检测的开发成本对大大都应用来说太高。连系亲和分子和量谱的卵白量组学检测不敷灵敏。生物和基因组学检测中高特异性的来源是分子校对。基于亲和分子的卵白量组学检测,包罗额外的校对步调,具有灵敏度和特异性的更佳组合,以及将来开展的更大潜力。概念总结卵白量组学检测必需具有足够的灵敏度和特异性,以便在其他卵白量存在的情况下对多种卵白量停止量化,那些卵白量可能以更高的浓度存在。基于量谱的卵白量组学检测不敷灵敏。基于抗体捕捉的卵白量组学检测既没有足够的特异性也没有足够的灵敏度。多重免疫夹心法检测的开发成本对大大都应用来说太高。连系亲和分子和量谱的卵白量组学检测不敷灵敏。生物和基因组学检测中高特异性的来源是分子校对。基于亲和分子的卵白量组学检测,包罗额外的校对步调,具有灵敏度和特异性的更佳组合,以及将来开展的更大潜力。概念总结卵白量组学检测必需具有足够的灵敏度和特异性,以便在其他卵白量存在的情况下对多种卵白量停止量化,那些卵白量可能以更高的浓度存在。基于量谱的卵白量组学检测不敷灵敏。基于抗体捕捉的卵白量组学检测既没有足够的特异性也没有足够的灵敏度。多重免疫夹心法检测的开发成本对大大都应用来说太高。连系亲和分子和量谱的卵白量组学检测不敷灵敏。生物和基因组学检测中高特异性的来源是分子校对。基于亲和分子的卵白量组学检测,包罗额外的校对步调,具有灵敏度和特异性的更佳组合,以及将来开展的更大潜力。概念总结卵白量组学检测必需具有足够的灵敏度和特异性,以便在其他卵白量存在的情况下对多种卵白量停止量化,那些卵白量可能以更高的浓度存在。基于量谱的卵白量组学检测不敷灵敏。基于抗体捕捉的卵白量组学检测既没有足够的特异性也没有足够的灵敏度。多重免疫夹心法检测的开发成本对大大都应用来说太高。连系亲和分子和量谱的卵白量组学检测不敷灵敏。生物和基因组学检测中高特异性的来源是分子校对。基于亲和分子的卵白量组学检测,包罗额外的校对步调,具有灵敏度和特异性的更佳组合,以及将来开展的更大潜力。概念总结卵白量组学检测必需具有足够的灵敏度和特异性,以便在其他卵白量存在的情况下对多种卵白量停止量化,那些卵白量可能以更高的浓度存在。基于量谱的卵白量组学检测不敷灵敏。基于抗体捕捉的卵白量组学检测既没有足够的特异性也没有足够的灵敏度。多重免疫夹心法检测的开发成本对大大都应用来说太高。连系亲和分子和量谱的卵白量组学检测不敷灵敏。生物和基因组学检测中高特异性的来源是分子校对。基于亲和分子的卵白量组学检测,包罗额外的校对步调,具有灵敏度和特异性的更佳组合,以及将来开展的更大潜力。概念总结卵白量组学检测必需具有足够的灵敏度和特异性,以便在其他卵白量存在的情况下对多种卵白量停止量化,那些卵白量可能以更高的浓度存在。基于量谱的卵白量组学检测不敷灵敏。基于抗体捕捉的卵白量组学检测既没有足够的特异性也没有足够的灵敏度。多重免疫夹心法检测的开发成本对大大都应用来说太高。连系亲和分子和量谱的卵白量组学检测不敷灵敏。生物和基因组学检测中高特异性的来源是分子校对。基于亲和分子的卵白量组学检测,包罗额外的校对步调,具有灵敏度和特异性的更佳组合,以及将来开展的更大潜力。诊断科学编纂团队搜集、整理和编撰,如需更多资讯,请存眷公家号诊断科学(DiagnosticsScience)。
参考文献
Anderson NL, Anderson NG. The human pla**a proteome: history, character, and diagnostic prospects. Mol. Cell Proteomics 1(11), 845–867 (2002). Anderson NL, Polanski M, Pieper R et al. The human pla**a proteome: a nonredun-dant list developed by combination of four separate sources. Mol. Cell Proteomics 3(4), 311–326 (2004). Sachdeva N, Asthana D. Cytokine quantitation: technologies and applications. Front Bio. Sci. 12, 4682–4695 (2007). Huang RP, Burkholder B, Jones VS et al. Cytokine antibody arrays in biomarker discovery and validation. Curr. Proteomics 9, 55–70 (2012). Saah AJ, Hoover DR. ‘Sensitivity’ and ‘specificity’ reconsidered: the meaning of these terms in ****ytical and diagnostic settings. Ann. Intern. Med. 126(1), 91–94 (1997). Pan S, Aebersold R, Chen R et al. Mass spectrometry based targeted protein quantification: methods and applications. J. Proteome Res. 8, 787–797 (2009). Mallick P, Kuster B. Proteomics: a pragmatic perspective. Nat. Biotechnol. 28, 695–709 (2010). Angel TE, Aryal UK, Hengel ** et al. Mass spectrometry-based proteomics: existing capabilities and future directions. Chem. Soc. Revs. 41, 39123928 (2012). Issaq HJ. The role of separation science in proteomics research. Electrophoresis 22, 3629–3638 (2001). Shi T, Fillmoreb TL, Sun X et al. Antibody-free, targeted mass-spectrometric approach for quantification of proteins at low picogram per milliliter levels in human pla**a/serum. Proc. Natl Acad. Sci. USA 109, 15395–15400 (2012). Fang X, Zhang WW. Affinity separation and enrichment methods in proteomic ****ysis. J. Proteomics 71(3), 284–303 (2008). Ekins R. The future development of immunoassays. In: Radioimmunoassay and Related Procedures in Medicine (Volume 1). Hagan AK, Zuchner T. Lanthanide-based time-resolved luminescence immunoassays. ****. Bio****. Chem. 400, 2847–2864 (2011). Natrajan A, Sharpe D, Costello J et al. Enhanced immunoassay sensitivity using chemiluminescent acridinium esters with increased light output. ****. Biochem. 406, 204–213 (2010). Sánchez-Carbayo M, Mauri M, Alfayate R, Miralles C, Soria F. ****ytical and clinical evaluation of TSH and thyroid hormones by electrochemiluminescent immunoassays. Clin. Biochem. 32(6), 395–403 (1999). MacBeath G. Protein microarrays and proteomics. Nat. Genet. 32(Suppl.), 526–532 (2002). Angenendt P. Progress in protein and antibody microarray technology. Drug Discov. Today 10(7), 503–511 (2005). King**ore SF. Multiplexed protein measurement: technologies and applications of protein and antibody arrays. Nat. Rev. Drug Discov. 5(4), 310–320 (2006). Chandra H, Reddy PJ, Srivastava S. Protein microarrays and novel detection platforms. Expert Rev. Proteomic 8, 61–79 (2011). Wilson R, Cossins AR, Spiller DG. Encoded microcarriers for high-throughput multiplexed detection. Angew. Chemie Int. Edit. 45, 6104–6117 (2006). Krishhan W, Khan IH, Luciw PA. Multiplexed microbead immunoassays by flow cytometry for molecular profiling: basic concepts and proteomics applications. Crit. Rev. Biotechnol. 29, 29–43 (2009). Huber W. Basic calculations about the limit of detection and its optimal determination. Accredit. Qual. Assur. 8, 213–217 (2003). Jackson TM, Ekins RP. Theoretical limitations on immunoassay sensitivity. Current practice and potential advantages of fluorescent Eu3+ chelates as non-radioiso-topic tracers. J. Immunol. Methods 87(1), 13–20 (1986). McConway MG, Chapman RS, Beastall GH et al. How sensitive are immunoassays for thyrotropin. Clin. Chem. 35, 289–291 (1989). Ekins RP. Immunoassay design and optimization. In: Principles and Practice of Immunoassay. Price CP, Newman DJ (Eds). Macmillan, London, UK, 175–207 (1997). Wingren C, Borrebaeck CA. Antibody microarray ****ysis of directly labelled complex proteomes. Curr. Opin. Biotechnol. 19(1), 55–61 (2008). Borrebaeck CA, Wingren C. Design of high-density antibody microarrays for disease proteomics: key technological issues. J. Proteomics 72(6), 928–935 (2009). Borrebaeck CA, Wingren C. High-through-put proteomics using antibody microarrays: an update. Expert Rev. Mol. Diagn. 7(5), 673–686 (2007). Kusnezow W, Syagailo YV, Goychuk I et al. Antibody microarrays: the crucial impact of mass transport on assay kinetics and sensitiv-ity. Expert Rev. Mol. Diagn. 6, 111–124 (2006). Wingren C, Ingvarsson J, Dexlin L, Szul D, Borrebaeck CA. Design of recombinant antibody microarrays for complex proteome ****ysis: choice of sample labeling-tag and solid support. Proteomics 7(17), 3055–3065 (2007). Ingvarsson J, Larsson A, Sj.holm AG et al. Design of recombinant antibody microar-rays for serum protein profiling: targeting of complement proteins. J. Proteome Res. 6(9), 3527–3536 (2007). Gao WM, Kuick R, Orchekowski RP et al. Distinctive serum protein profiles involving abundant proteins in lung cancer patients based upon antibody microarray ****ysis. BMC Cancer 5, 110 (2005). Orchekowski R, Hamelinck D, Li L et al. Antibody microarray profiling reveals individual and combined serum proteins associated with pancreatic cancer. Cancer Res. 65(23), 11193–11202 (2005). Sanchez-Carbayo M, Socci ND, Lozano JJ, Haab BB, Cordon-Cardo C. Profiling bladder cancer using targeted antibody arrays. Am. J. Pathol. 168(1), 93–103 (2006). Shafer MW, Mangold L, Partin AW, Haab BB. Antibody array profiling reveals serum TSP-1 as a marker to distinguish benign from malignant prostatic disease. Prostate 67(3), 255–267 (2007). Loch CM, Ramirez AB, Liu Y et al. Use of high density antibody arrays to validate and discover cancer serum biomarkers. Mol. Oncol. 1(3), 313–320 (2007). Haab BB, Dunham MJ, Brown PO. Protein microarrays for highly parallel detection and quantitation of specific proteins and antibodies in complex solutions. Genome Biol. 2(2), RESEARCH0004 (2001). Michaud GA, Salcius M, Zhou F et al. ****yzing antibody specificity with whole proteome microarrays. Nat. Biotechnol. 21(12), 1509–1512 (2003). Kijanka G, Ipcho S, Baars S et al. Rapid characterization of binding specificity and cross-reactivity of antibodies using recombinant human protein arrays. J. Immunol. Methods 340(2), 132–137 (2009). Whiteaker JR, Zhao L, Anderson L, Paulovich AG. An automated and multiplexed method for high throughput peptide immunoaffinity enrichment and multiple reaction monitoring mass spectrometry-based quantification of protein biomarkers. Mol. Cell Proteomics 9(1), 184–196 (2010). Pontén F, Schwenk JM, Asplund A, Edqvist PH. The Human Protein Atlas as a proteomic resource for biomarker discovery. J. Intern. Med. 270(5), 428–446 (2011). Schwenk JM, Igel U, Neiman M et al. Toward next generation pla**a profiling via heat-induced epitope retrieval and array-based assays. Mol. Cell Proteomics 9(11), 2497–2507 (2010). Dübel S, Stoevesandt O, Taussig MJ, Hust M. Generating recombinant antibodies to the complete human proteome. Trends Biotechnol. 28(7), 333–339 (2010). Bradbury AR, Sidhu S, Dübel S, McCafferty J. Beyond natural antibodies: the power of in vitro display technologies. Nat. Biotech-nol. 29(3), 245–254 (2011). Levin AM, Weiss GA. Optimizing the affinity and specificity of proteins with molecular display. Mol. Biosyst. 2(1), 49–57 (2006). Altshuler EP, Serebryanaya DV, Katrukha AG. Generation of recombinant antibodies and means for increasing their affinity. Biochemistry Mosc. 75(13), 1584–1605 (2010). Markus V, Janne L, Urpo L. Directed antibody-engineering techniques and their applications in food immunoassays. Trac. Trend ****. Chem. 30, 219–226 (2011). Ragoussis J. Genotyping technologies for genetic research. Annu. Rev. Genomics Hum. Genet. 10, 117–133 (2009). LaFratta CN, Walt DR. Very high density sensing arrays. Chem. Rev. 108(2), 614–637 (2008). Shen R, Fan JB, Campbell D et al. High-throughput SNP genotyping on universal bead arrays. Mutat. Res. 573(1–2), 70–82 (2005). Hopfield JJ. Kinetic proofreading: a new mechani** for reducing errors in biosyn-thetic processes requiring high specificity. Proc. Natl Acad. Sci. USA 71(10), 4135–4139 (1974). Wu WW, Slastad H, Carillo DD et al. Antibody array ****ysis with label-based detection and resolution of protein size. Mol. Cell. Proteomics 8, 245–257 (2009). Weibrecht I, Leuchowius KJ, Clausson CM et al. Proximity ligation assays: a recent addition to the proteomics toolbox. Expert Rev. Proteomics 7(3), 401–409 (2010). Landegren U, V.nelid J, Hammond M et al. Opportunities for sensitive pla**a proteome ****ysis. ****. Chem. 84(4), 1824–1830 (2012). Lundberg M, Thorsen **, Assarsson E et al. Multiplexed homogeneous proximity ligation assays for high-throughput protein biomarker research in serological material. Mol. Cell. Proteomics 10, 004978 (2011). Kim J, Hu J, Sollie RS, Easley CJ. Improvement of sensitivity and dynamic range in proximity ligation assays by asymmetric connector hybridization. ****. Chem. 82(16), 6976–6982 (2010). Darmanis S, Nong RY, Hammond M et al. Sensitive pla**a protein ****ysis by microparticle-based proximity ligation assays. Mol. Cell Proteomics 9(2), 327–335 (2010). S.derberg O, Leuchowius KJ, Gullberg M et al. Characterizing proteins and their interactions in cells and tissues using the in situ proximity ligation assay. Methods 45(3), 227–232 (2008). Fredriksson S, Gullberg M, Jarvius J et al. Protein detection using proximity-depend-ent DNA ligation assays. Nat. Biotechnol. 20(5), 473–477 (2002). Stoltenburg R, Reinemann C, Strehlitz B. SELEX – a (r)evolutionary method to gener-ate high-affinity nucleic acid ligands. Biomol. Eng. 24(4), 381–403 (2007). Song KM, Lee S, Ban C. Aptamers and their biological applications. Sensors (Basel) 12(1), 612–631 (2012). Wilson R, Cossins A, Nicolau DV, Missailidis S. The selection of DNA aptamers for two different epitopes of thrombin was not due to different partitioning methods. Nucleic Acid Ther. 23(1), 88–92 (2013). Gold L, Ayers D, Bertino J et al. Aptamer-based multiplexed proteomic technology for biomarker discovery. PLoS ONE 5(12), e15004 (2010). Wilson R. High-content aptamer-based proteomics. J. Proteomics 74(10), 1852–1854 (2011). Mehan MR, Ayers D, Thirstrup D et al. Protein signature of lung cancer tissues. PLoS ONE 7(4), e35157 (2012). Editors Profile. Lee Hood. Nat. Biotechnol. 29, 191 (2011). Fan R, Vermesh O, Srivastava A et al. Integrated barcode chips for rapid, multiplexed ****ysis of proteins in microliter quantities of blood. Nat. Biotechnol. 26(12), 1373–1378 (2008). Gervais L, de Rooij N, Delamarche E. Microfluidic chips for point-of-care immunodiagnostics. Adv. Mater. Weinheim 23(24), H151–H176 (2011). Ruigrok VJ, Levisson M, Eppink MH, **idt H, van der Oost J. Alternative affinity tools: more attractive than antibodies? Biochem. J. 436(1), 1–13 (2011). Gold L, Walker JJ, Wilcox SK, Williams S. Advances in human proteomics at high scale with the SOMAscan proteomics platform. N. Biotechnol. 29(5), 543–549 (2012). Vaught JD, Bock C, Carter J et al. Expanding the chemistry of DNA for in vitro selection. J. Am. Chem. Soc. 132(12), 4141–4151 (2010). Pinheiro VB, Holliger P. The XNA world: progress towards replication and evolution of synthetic genetic polymers. Curr. Opin. Chem. Biol. 16(3–4), 245–252 (2012). Eaton BE. The joys of in vitro selection: chemically dressing oligonucleotides to satiate protein targets. Curr. Opin. Chem. Biol. 1(1), 10–16 (1997). Nong RY, Gu J, Darmanis S, Kamali-Moghaddam M, Landegren U. DNA-assist-ed protein detection technologies. Expert Rev. Proteomics 9(1), 21–32 (2012). Müller UR. Protein detection using biobarcodes. Mol. Biosyst. 2(10), 470–476 (2006).***